- Lupe_Grau Cancel

In Vitro Diagnostic Medical Device Performance Evaluation: 8 Steps to Conformity
Tuesday, May 5, 2020
If manufacturers don’t conduct a legally compliant performance evaluation of their in vitro diagnostic medical device (IVD) , they aren’t just running the risk of problems during the authorization process.
They are risking patient safety. Therefore, the IVDR sets strict requirements for this performance evaluation.
Find out how the requirements of the IVDR for the performance evaluation differ from those of the IVDD to make sure you achieve conformity as quickly as possible .
The most common mistakes can be avoided in eight steps.
1. Why the IVD performance evaluation is so important
Summary
In this section, you will learn about the potential negative consequences of a poor performance evaluation. As an IVD manufacturer, you should make absolutely sure to avoid them.
a) The role of in vitro diagnostic medical devices
The purpose of in vitro diagnostic medical devices (IVDs) is to provide information from human samples, such as blood and tissue, that allows conclusions to be drawn about, for example, physiological or pathological processes in the body.
An IVD is used to identify, for example:
- Tumor markers in blood
- Coronavirus (SARS-CoV-2) in a smear
- Cancer cells in a biopsy
This information is crucial for making a diagnosis and, therefore, for the further treatment of a patient. The IVD performance evaluation must ensure that the information provided is correct and accurate, and that it provides the intended clinical benefit.
b) Potential IVD errors
However, the information generated using an IVD can be incorrect in several respects:
- False positive : A laboratory test incorrectly says that a test result is abnormal or detects a disease even though the patient is not ill.
- False negative : A laboratory test incorrectly says that a test result is not abnormal or does not detect a disease even though the patient is ill.
- The information is inaccurate .
- The IVD does not display the information at all or only displays it with a delay .
There are numerous reasons for such errors. Examples are:
- The analysis procedure is unsuitable in general.
- The manufacturer has not considered all the factors that could affect the tissue samples, e.g., the effect of drugs.
- Reagents change, for example, under the influence of heat, oxygen or light.
- Software errors cause patients to get mixed up.
- When the samples are opened, even the smallest splashes lead to cross-contamination of other samples.
c) Consequences for patients
There is a risk that incorrect information could have far-reaching consequences for patients.
False positive results generally trigger an unnecessary cascade of measures that harm the patient, or at least cause them unnecessary stress:
- Unnecessary blood draws
- Avoidable biopsies
- Wrong therapies, e.g., with medicines or even operations
False negative results, in contrast can lead to critical delays to urgently needed therapies or even have an impact on public health.
d) Consequences for manufacturers
The performance evaluation doesn’t just enable IVD manufacturers to ensure that the risks for patients have been minimized, it also enables them to ensure that the consequences for their own company have been minimized as well.
- The device “fails the authorization” and is therefore released onto the market later. As a result, the company loses planned sales .
- The device has to be withdrawn from the market. This doesn’t just have financial implications for the company, it also damages their reputation . Manufacturers have to publish such recalls on the websites of the corresponding authorities.
- In the worst case, manufacturers will be faced with claims for compensation from patients harmed by the device.
- In addition to unsatisfied customers and a damaged reputation, manufacturers will also face additional costs for support and improvements .
2. What a performance evaluation is and what it has to demonstrate
In this section, you will be introduced to important concepts and definitions that you will need to know in order to understand the legal texts.
a) Definitions
Fortunately, the IVDR contains several definitions:
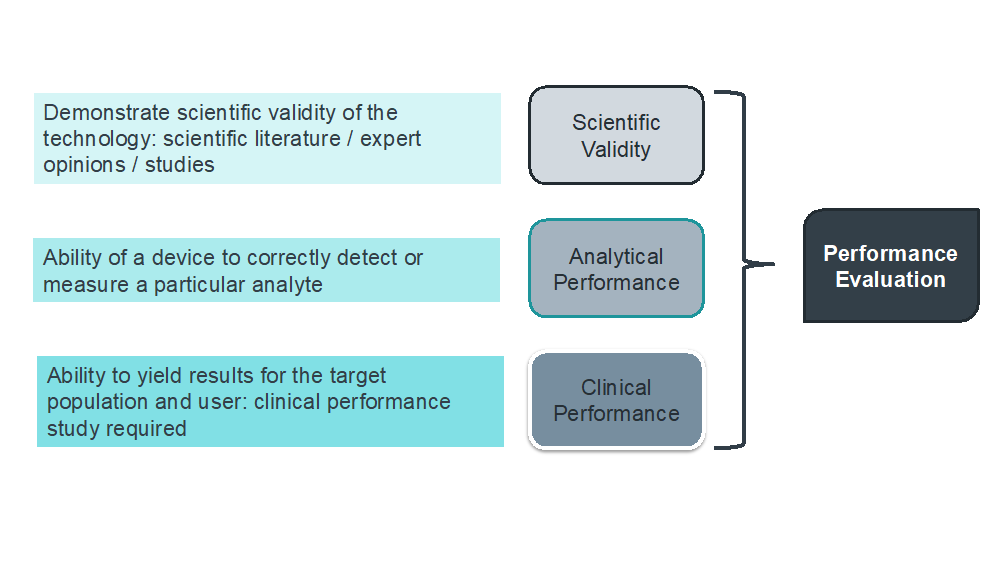
Definition: Performance evaluation
“Assessment and analysis of data to establish or verify the scientific validity, the analytical and, where applicable, the clinical performance of a device”
IVDR Article 2(44)
This definition uses other defined terms:
- Scientific validity
- Analytical performance
- Clinical performance
Definition: Scientific validity of an analyte
“Association of an analyte with a clinical condition or a physiological state”
IVDR Article 2(38)
The basis for each IVD is the analyte that is used to identify a physiological state or disease. For example, there is a link between the coronavirus (SARS-CoV-2) and the disease Covid-19. Similarly, there is a link between the level of prostate-specific antigen (PSA) in the blood and the risk of a man suffering from prostate cancer.
Definition: Analytical performance
“Means the ability of a device to correctly detect or measure a particular analyte”
IVDR Article 2(40)
To ensure that the analyte can also be reliably measured using the method chosen, IVD manufacturers should check the analytical performance of their device. To continue with the examples: The analytical performance of an IVD is its ability to identify the coronavirus as accurately as possible (i.e., without false-positive or false-negative results) or to determine the PSA concentration as accurately as possible.
Definition: Performance of a device
“Ability of a device to achieve its intended purpose as claimed by the manufacturer. It consists of the analytical and, where applicable, the clinical performance supporting that intended purpose”
IVDR Article 2(39)
This definition uses another term, “clinical performance,” that we need to understand.
Definition: Clinical performance
“Ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user”
IVDR Article 2(41)
b) Clinical evidence
IVD manufacturers must provide proof of an IVD’s clinical benefit based on data on scientific validity, analytical performance and clinical performance. Scientific validity refers to the association between the analyte and the disease or physiological state. After all, for the IVD to deliver clinical performance, it must have a reliable and precise method for measuring the analyte. Such a method demonstrates the ability of the IVD to determine clinical, physiological and pathological states or identify processes, and is therefore a vital aspect of the device's performance.

It is also important to note that clinical performance does not refer to an absolute ability but, in fact, depends on the intended purpose and thus the “target population” and “intended user.”
The clinical performance of an IVD may be good for “normal” patients but not for patients undergoing chemotherapy because the accuracy of its measurement is affected by cytostatics.
A device's performance may be excellent for professional users, but not for laypersons. Therefore, the IVDR stresses that an IVD’s performance depends on its intended purpose.
The clinical evidence, i.e., the proof that a device is safe and achieves the intended clinical benefit, is provided by the performance evaluation . This performance evaluation assesses clinical performance, analytical performance, and scientific validity.
3. Which performance evaluation regulatory requirements do IVD manufacturers have to comply with?
This section will give you an overview of
- The relevant regulations (without having to research them first)
- The most important requirements
You will also find out what deltas there are between the IVDD and IVDR that you may need to close.
Both the IVD Directive 98/79/EC (IVDD) and the IVD Regulation 2017/746 (IVDR) establish, in their respective Annex Is, that the general safety and performance requirements must be complied with. Demonstrating this compliance is part of the performance evaluation.
The IVDR states that in order for a device to be placed on the market or put into service:
“A device shall meet the general safety and performance requirements set out in Annex I which apply to it, taking into account its intended purpose. [...] Demonstration of conformity with the general safety and performance requirements shall include a performance evaluation.”
Article 5, IVDR
Article 56 requires manufacturers to confirm that the general safety and performance requirements according to Annex I have been met. Manufacturers also have to carry out a performance evaluation to provide this confirmation.
This performance evaluation includes (as outlined above) the demonstration of the scientific validity of the analyte, the analytical performance and the clinical performance.
The focus is on demonstrating the following in particular:
- The general requirements set out in Annex I Chapter I
- The performance characteristics set out in Annex I, paragraph 9
- An acceptable benefit-risk ratio
The article refers to the requirements established in Annex XIII, Part A with regard to the performance evaluation.
Annexes XIII and ISO 20916:2019
This annex specifies how IVD manufacturers must plan, carry out and document the performance evaluation. Manufacturers should also refer to ISO 20916:2019, which the amended IVDR of May 03, 2019 directly references.
“ The rules on performance studies should be in line with well-established international guidance in this field, such as the international standard ISO 20916 on clinical performance studies using specimens from human subjects, currently under development, so as to make it easier for the results of performance studies ... ”
IVDR, Recital 66
ISO 20916:2019 was published in May 2019. Its title is “In vitro diagnostic medical devices – Clinical performance studies using specimens from human subjects – Good study practice .”
ISO 20916:2019 describes in detail the requirements for the planning and performance of clinical performance studies. It supplements the IVDR with regard to the description of the organization, the roles involved and the requirements for conducting such a study.
There are overlaps with the IVDR, particular with regard to the content of the clinical performance study plan (according to IVDR, Annex XIII, paragraph 2.3.2). ISO 20916 calls this plan the “ clinical performance study protocol (CPSP) .”
Annexes A to F of ISO 20916:2019 are dedicated to the specific requirements for interventional and other types of performance studies in accordance with Article 57 of the IVDR. The IVDR itself provides detailed specifications for these studies in Annex XIV.
Annex II: Technical documentation
Annex II describes the technical documentation requirements. In paragraph 6 of this annex, the IVDR describes what the performance evaluation, verification and validation should contain.
Annex VII: A look behind the facade
Annex VII describes the requirements to be met by notified bodies. Understanding what notified bodies have to look for during conformity assessment activities helps us to work out what is important in the performance evaluation and where the interfaces are.
Focus of the assessments
According to Annex VII, paragraph 4.5.4 of the IVDR, the focus of the assessment should be on a manufacturer’s procedures and the documentation regarding:
- The planning, conduct, assessment, reporting and updating of the performance evaluation
- Post-market surveillance and post-market performance follow up (PMPF)
- The interface with the risk management process
- The appraisal and analysis of the available data and its relevance with regard to demonstrating conformity with the general safety and performance requirements set out in Annex I of the IVDR
- The performance evaluation report
The annex explicitly requires notified bodies to take into consideration the available common specifications , guidance and best practice documents.
Literature search
They must also assess the results of the literature searches, of the verifications and validations performed, and of other tests. Notified bodies must also assess the packaging, stability studies, and results of the shelf life tests.
The assessment of the performance evaluation must expressly cover:
- The intended purpose and intended use
- The planning of the performance evaluation
- The methodology for the literature search and its documentation
- The analytical and clinical performance studies
- Justifications in relation to non-performance of performance studies or deviations from the specified process
- If data from devices considered equivalent are used for the performance evaluation, the validity of the assumed equivalence and the suitability of the data for demonstrating conformity must be verified
It is noteworthy that the notified body must ensure:
- “That the conclusions drawn by the manufacturer are valid in the light of the approved performance study plan”
- That “the performance evaluation adequately addresses the relevant safety and performance requirements provided for in Annex I”
- That the performance evaluation is “appropriately aligned with the risk management requirements”
- That the performance evaluation “is conducted in accordance with Annex XIII”
- The performance evaluation “is appropriately reflected in the information provided relating to the device”
This gives us hope that there will be significantly more transparency and comparability in future when it comes to information on an IVD’s performance.
b) MPG, IVDD and EN 13612:2012
The requirements of the MPG (German Medical Devices Act) refer to the In Vitro Diagnostics Directive (IVDD). Here too the focus is on the intended purpose. And here too, IVDs must – taking into account their intended purpose – meet the relevant essential requirements according to Annex I. (see MPG § 6 , paragraph 2).
“ Evidence of the suitability of in vitro diagnostic medical devices for the specified in-tended purpose should be provided through performance evaluation based on appropriate data. The performance evaluation should be based on: – data from scientific literature which cover the intended use of the medical device and the techniques involved in its use as well as a written report containing a critical evaluation of these data or – the results of all performance evaluation studies or other appropriate tests .”
MPG, Section 19, paragraph 2
In Annex I, paragraph 3 the IVDD lists the essential performance parameters that an IVD must meet (unless the parameters can be excluded with good reason). Furthermore, in Annex I, paragraphs 1 and 2, the directive describes the requirements for the safety of the device and the acceptability of the benefit-risk ratio.
In the third paragraph of Annex III, indent 11 requires “ adequate performance evaluation data showing the performances claimed by the manufacturer and supported by a reference measurement system .”
The standard EN 13612:2002 is harmonized under the IVDD. The standard, which is entitled “ Performance evaluation of in vitro diagnostic medical devices ” specifies “ the responsibilities and general requirements for the planning, conduct, assessment and documentation of a performance evaluation study .”
c) IMDRF/GHTF
Guidance documents reflect the state of the art.
Further information of conducting and documenting performance evaluations can be found in the guidance documents from the IMDRF (formerly the GHTF). They represent the current state of the art under the IVDD and were the basis for the procedure as it is now described in the IVDR.
In some places you can find word-for-word the same requirements. Therefore, the requirements of the IVDR are – contrary to what a lot of manufacturers have claimed – not new at all, rather they have been the state of the art since 2012.
Applicable documents
Manufacturers should pay particular attention to the following documents:
- GHTF/SG5/N6:2012 “Clinical Evidence for IVD medical devices – Key Definitions and Concepts” Since 2012, this document has defined the terms that we now find in the IVDR. This includes, e.g., clinical evidence, scientific validity, analytical performance, clinical performance, and clinical utility (clinical benefit). Section 4.10 even provides an example of how to provide clinical evidence for an IVD.
- GHTF/SG5/N7:2012 “Clinical Evidence for IVD medical devices – Scientific Validity Determination and Performance Evaluation” This document looks at the underlying process for presenting clinical evidence in more detail. The procedures for determining the scientific validity of the analyte, verifying analytical performance and validating the clinical performance are described step by step. The requirements for a systematic literature search (see Annex B) and documenting in it a literature search report (see Appendix A) are also described.
- GHTF/SG5/N8:2012 “Clinical Evidence for IVD Medical Devices – Clinical Performance Studies for In Vitro Diagnostic Medical Devices” This guidance document now goes into detail on the requirements for IVD clinical performance studies. It describes the different types of studies and explains the numerous that have to be considered when planning a clinical performance study. Further chapters outline the content of the CPSP and CPSR and specify the requirements for the conduct of the clinical performance study.
4. A performance evaluation in 8 steps – what you should do as manufacturer
In this section, we will explain step-by-step how you can conduct target-oriented legally compliant performance evaluations.
Step 1: Create and review a standard operating procedure
First write a standard operating procedure for IVD performance evaluations and integrate it into your QM system. How to create a standard operating procedure is described below.
Make sure that the standard operating procedure covers the entire IVD performance evaluation process. It should cover the complete life cycle of the device and goes far beyond development.
Note : Although ISO 13485 does not explicitly require a process for the performance evaluation to be defined, it does require, in section 4.2.1 e), all other documentation specified by applicable regulatory requirements. And this requirement is found in the IVDR in Article 56 and Annex XIII, among others. The IVDR describes the performance evaluation as a continuous process . It emphasizes the close interface with risk management (Annex VII, Section 4.5.4 and Annex XIII section 1.1).
Step 2: Write a device-specific performance evaluation plan
The next step is to write a specific performance evaluation plan for each IVD. Make sure that you include all the content required by Annex XIII, paragraph 1.1 of the IVDR in the performance evaluation plan.
In the performance evaluation plan, you should describe the current state of the art , with regard to the medically or diagnostically relevant field as well as in terms of the technology.
Research in guidelines, the scientific literature and standards, and document this literature search clearly.
Please note:
- As an IVD manufacturer, you can derive the device requirements (“Design and development inputs” according to ISO 13485, 7.3.3) and the device design specifications (“Design and development outputs” according to ISO 13485, 7.3.4) from the results of the literature search.
- The research into the state of the art will generate results (e.g., about the benefits and risks of comparable devices) that you will also need for risk management.
- Create the performance evaluation plan at the start of the IVD development stage. It is common practice to continuously add to this plan when new findings become known.
Step 3: Demonstrate scientific validity
Now development can start. Demonstrate the scientific validity of the analyte(s) to be detected or measured by the IVD as early as possible in the development cycle.
Carry out a further systematic literature search . Document this search clearly, e.g., the search strategy, the sources used and the criteria for selecting the data. Statements by professional societies and expert opinions can also be included.
If this evidence is not sufficient, carry out feasibility studies . Their results can be used to prove the scientific validity of the analyte, particularly in the case of novel analytes.
Summarize all the results in a scientific validity report . Add the report to the performance evaluation file, which is itself part of the technical documentation .
Note : Use your literature search report in the continuous process to update the performance evaluation and during the post-market performance follow-up (PMPF).
Step 4: Conduct the analytical performance evaluation
Now you can start with the analytical performance evaluation as part of the IVD verification process. Describe your planned procedure and the concrete experimental design in an analytical performance evaluation plan ( verification plan ).
If you have a complex IVD system (e.g., consisting of an IVD device, IVD assay and IVD software), plan the verification of
- The individual subsystems
- Their integration
- The overall system
As well as the analysis itself, you should also plan the sampling and sample handling precisely because reproducible and accurate analysis results can only be obtained if the conditions during the sampling and preparation of the samples are controlled.
Then conduct the evaluation according to the plan and finally summarize the results of the analytical performance studies in an analytical performance report . Please also evaluate the results in this report.
- The demonstration of analytical performance must always be based on analytical performance studies.
- The analytical performance evaluation, as part of the verification, is used to demonstrate the analytical performance parameters according to Annex I, paragraph 9.1 a) of the IVDR.
Step 5: Conduct the clinical performance evaluation
Next move on to the clinical performance evaluation , which will show the diagnostic accuracy of your IVD.
- As a general rule, clinical performance study results are required to assess the clinical performance characteristics. In justified exceptions, you can refer only to scientific literature (e.g., in the case of established, standardized tests and where the equivalence of the device has been demonstrated) or to published experiences gained from routine diagnostic tests (see the article on lab developed tests ).
- This basic strategy for demonstrating clinical performance is already described in the performance evaluation plan from step 2.
If you use (supplementary) literature data, document your systematic literature search clearly.
Write another plan for the clinical performance studies – the clinical performance study protocol (CPSP) . Make sure that you include the aspects required by Annex XIII, paragraph 2 of the IVDR as well as those required by ISO 20916:2019. You should also make sure that the plan provides for the collection of data for all parameters listed in Annex I, paragraph 9.1 b).
Finally, summarize the results of the clinical performance evaluation of your IVD in a clinical performance report .
Step 6: Demonstrate the stability of the IVD
Depending on the type of device you have, you now need to demonstrate its stability. This evidence is particularly important for IVD assays and in vitro diagnostic reagents.
First plan the stability studies. For this, follow the advice in the EP25 guidance document from the Clinical and Laboratory Standards Institute (CLSI) . When preparing your plan, make sure that you take all stability aspects mentioned by the IVDR in Annex II, paragraph 6.3 into account. These are in particular:
- The device’s shelf life
- The in-use stability (e.g., of the reagents)
- Transport stability
Step 7: Prepare the clinical performance evaluation report
Lastly, summarize all the results on scientific validity, on analytical performance and clinical performance in the performance evaluation report .
In the report, you should evaluate the clinical evidence in the light of the current state of the art in medicine and demonstrate your device's positive benefit-risk ratio.
Step 8: Create and implement a PMPF plan
Create a plan for the post-market performance follow-up plan (PMPF plan) . You should take into account the requirements of Annex XIII, Part B, paragraph 5.2 when doing so.
Note : PMPF is the acronym of the English term “post-market performance follow-up”.
Conduct this follow-up for your CE-marked and marketed device and summarize the results in the PMPF report. Use the results of this report to update the performance evaluation report.
Note : The performance evaluation is a continuous process. The performance evaluation activities are carried out during the device's entire life cycle. Therefore, manufacturers are obliged to continuously surveil their device and update the performance evaluation with new information and data that becomes available.
5. What are the common mistakes made during the performance evaluation?
This section will describe the biggest mistakes you should avoid. Use this list as a checklist to avoid unnecessary hassle and costs.
a) Unspecific intended purpose
Manufacturers often formulate the intended use in a more general and less specific way:
- No limitation of indication (all types of cancer)
- No limitation of target population
As a result, the performance also has to be demonstrated with no limitations on, e.g., indications and target populations. The scope of the studies required grows exponentially. Furthermore, requirements that apply, for example, for performance studies in children, pregnant or breastfeeding women (see Articles 61 and 62) are easily overlooked.
b) Unsystematically researched or documented state of the art
If manufacturers do not systematically research the state of the art and do not clearly document this research, there will be a lack of
- Reliable specifications of analytical and clinical performance data
- Knowledge and analysis of competitor devices
- User knowledge, the use environment and the intended clinical benefit (what does the physician or patient do when they have the information from the IVD? What actions and therapies are possible?)
Lack of knowledge of the state of the art leads to manufacturers not setting device requirements and, for example, not specifying sufficient controls for the device (e.g., self-tests).
c) No documented systematic literature review
If the literature review is not conducted in full and documented clearly, the manufacturer cannot demonstrate that they have met the performance requirements. Authorities and notified bodies will notice this and refuse authorization.
d) Inadequate plans for analytical performance evaluation
The typical mistakes include missing or incomplete performance evaluation plans. This leads, for example, to analytical performance studies without a statistically valid experimental design.
Reasons for inadequate plans include a lack of knowledge of the state of the art with regard to planning, which is described in the CLSI standards . Incidentally, the IVDD harmonized standard EN ISO 18113-1:2011 “ In vitro diagnostic medical devices – Information supplied by the manufacturer (labelling) – Part 1: Terms, definitions and general requirements ” refers to these CLSI standards.
e) Unsuitable performance studies
The following mistakes are often observed during clinical performance studies:
- There is no explanation of the number of cases or the number of samples.
- The performance study does not take the intended purpose into account sufficiently. As a result, the study design is not suitable.
- The manufacturer does not derive the clinical performance parameters from the state of the art specifically for the promised clinical benefit. As a result, the performance studies do not have the correct endpoints.
f) No performance data from studies
Manufacturers often hope to get by without clinical performance data from studies. This is possible in principle. However, it requires equivalence between the IVD to be evaluated and the comparator device. This technical and medical or diagnostic equivalence must be analyzed in detail and the evidence documented. Unfortunately, the information required is often missing.
g) Lack of proof of scientific validity
It is not enough to just demonstrate analytical performance. Manufacturers must also prove the scientific validity of the analyte. This is forgotten because the IVDD does not explicitly require this, even though it is the state of the art according to GHTF/SG5/N7:2012.
6. Support from the Johner Institute
The IVD experts at the Johner Institute can provide support to IVD manufacturers with all performance evaluation activities and phases:
- Strategy development
- Determination of the state of the art
- Planning and conduct of the PMPF activities and studies, and updating of the performance evaluation
The Johner Institute’s IVD team can
- Help you create standard operating procedures for the performance evaluation
- Carry out the literature search for you and help you make sure your documentation is clear and comprehensible
- Create performance evaluation plans and reports for you
- Provide support for all types of study
- Review and, if requested, correct your files before submission
- Hold on-site or online seminars, training courses and workshops
Get in touch if you want to conduct a quick and legally compliant performance evaluation that will be waved through by your notified body with no problems.
7. Conclusion & summary
A) performance evaluation aspects.
The performance evaluation is as important for IVD manufacturers as the clinical evaluation is for medical device manufacturers. Both are based on data that are already available in the literature or that have to be collected through studies.
The nature of the studies and the data differ considerably. When evaluating the performance of IVDs, manufacturers must demonstrate:
Each of these aspects requires its own plans, data and evaluations.
b) Comparison of IVDD and IVDR
The performance evaluation requirements established in the IVDR and the standard ISO 20916:2019 it references are significantly more wide-ranging than those established by the IVDD. However, under the IVDD, the GHTF and CLSI documents and EN 13612:2002 were already considered the state of the art. So, there aren’t any significant differences.
This means that IVD manufacturers who already comply with the relevant regulations are well prepared for the IVDR. However, a lot of IVDs will be subject to a conformity assessment by a notified body for the first time under the IVDR. All IVD manufacturers should prepare well for the detailed review of the performance evaluation file.
c) MDR and IVDR challenge?
The situation is challenging for manufacturers who must comply with the IVDR’s requirements for the performance evaluation and the MDR's requirements for the clinical evaluation because their devices are subject to both regulations. However, this can be avoided with a clever regulatory strategy.
Do you have any questions about this article? Contact us via our micro-consulting team. The Johner Institute’s IVD team will be happy to help.

Dr. Catharina Bertram

A quick overview: Our
Starter-Kit
Always up to date: Our
Back To Top
Privacy settings
We use cookies on our website. Some of them are essential, while others help us improve this website and your experience.
Individual Cookie Settings
Only accept required cookies.
Privacy Notes Imprint
Here is an overview of all cookies use
Required Cookies
These cookies are needed to let the basic page functionallity work correctly.
Show Cookie Informationen
Hide Cookie Information
Provide load balancing functionality.
Provides functions across pages.
Hubspot Forms
Used for the google recaptcha verification for online forms.
Cookies for Statistics
Statistic cookies anonymize your data and use it. These information will help us to learn, how the users are using our website.
Google Analytics
Tracking and analys of traffic on our websites.
Cookies for Marketing
Marketing cookies from thrid parties will be used to show personal advertisment. They use them to track users outside of their own web page.
Keeping track of a visitor's identity. It is passed to HubSpot on form submission and used when deduplicating contacts. It contains an opaque GUID to represent the current visitor. It also introduces cookies from linked in for marketing reasons.
LinkedIn conversion tracking.
Cookies for external Content
Content for Videoplatforms und Social Media Platforms will be disabled automaticly. To see content from external sources, you need to enable it in the cookie settings.
Google Maps
Used to display google maps on our Websites. Google uses cookies to identify and track users.

Guest Column | August 25, 2021
Best practices for writing an ivdr-compliant performance evaluation report.
By Priya Ray Chaudhuri, Freyr Solutions

A legally non-compliant performance evaluation of an in-vitro diagnostic medical device (IVD) not only poses a risk of problems with the product during the authorization process but also risks patient safety. This paved the path for strict and robust IVDR requirements on performance evaluation of IVD products.
What Is Clinical Evidence?
As per the new European regulation 2017/746 on in vitro diagnostic devices (the EU-IVDR), “’Clinical evidence’ means clinical data and performance evaluation results, … of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s) when used as intended by the manufacturer.” 1
In short, clinical evidence is the data that supports the use of the device, which is required for all IVDs irrespective of class, based on assessed data used to demonstrate compliance with the general safety and performance requirements (GSPRs) laid out in Annex I of the regulation. As the IVDs are classified in accordance with the rules set out in Annex VIII of the regulation in a risk-based approach, the amount and quality of the clinical data varies among the device classes.
How To Gather Clinical Data For An IVD
The building blocks of the clinical evidence are based on three integral pillars for an in-vitro diagnostic device, namely:
- Scientific validity
- Analytical performance
- Clinical performance
Scientific Validity : Scientific validity means the association of an analyte or marker with a clinical condition or a physiological state. This can be demonstrated through a literature search if enough information with adequate quality can be found to establish the validity. 2
Additionally, consensus expert opinions result from proof of concept, and clinical performance studies may be utilized as sources of data. The scientific validity of the analyte or marker is documented in the scientific validity report.
Analytical Performance: Analytical performance is the ability of a device to correctly detect or measure a particular analyte. The analytical performance of the device shall be demonstrated in relation to the following parameters (unless any of them can be justified as not applicable): analytical sensitivity, analytical specificity, trueness (bias), precision (repeatability and reproducibility), accuracy (resulting from trueness and precision), limits of detection and quantitation, measuring range, linearity, cut-off (including determination of appropriate criteria for specimen collection and handling and control of known relevant endogenous and exogenous interference), and cross-reactions.
Generally, analytical performance is demonstrated based on analytical performance studies and is demonstrated and documented in the analytical performance report.
Clinical Performance: Clinical performance is the ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user. The clinical performance of the device shall be demonstrated using the following parameters (unless any of them can be justified as not applicable): diagnostic sensitivity, diagnostic specificity, positive predictive value, negative predictive value, likelihood ratio, and expected values in normal and affected populations.
Demonstration of clinical performance must be based on the clinical performance studies, scientific peer-reviewed literature, and/or publishing experience gained by routine diagnostic testing. Clinical performance studies should always be performed unless it can be justified that a demonstration based on other sources of clinical data is sufficient. The clinical performance should be demonstrated and documented in the clinical performance report.
The clinical evidence gathered from these elements occurs throughout the lifetime of the device, which results in a rule of thumb as depicted below:

A performance evaluation plan or PEP consists of the procedures and methods to correctly perform and appropriately report the performance evaluation. According to the EU-IVDR, the PEP should cover at least the following:
Performance Evaluation Plan
- The intended purpose of the IVD
- Description of the analyte
- Target population
- Description of the state-of-the-art
- Steps for demonstrating the scientific validity, clinical performance, and analytical performance
- Determination of the acceptability of the benefit-risk ratio
Performance Evaluation Report (PER)
The performance evaluation report is an output of the process of performance evaluation activities populated from the results of applying the performance evaluation plan. Annex XIII, Part A (1.3.2) of the IVDR outlines the specific components of the PER and specifies that it must include:
- The scientific validity report
- The analytical performance report
- The clinical performance report and
- An assessment of all these reports supporting that the demonstration of the clinical evidence is sufficient to decide on the benefit-risk ratio.
Performance evaluation reports for Class C and D devices 3,4 must be updated at least annually, whereas PERs for Class A and B 5 devices should be updated as needed, although at least a three-year review cycle is recommended. Along with the above-mentioned elements of the performance evaluation, this should include continuous planning and gathering reports of post-market surveillance, as well as identifying and assessing any new/upcoming/residual risks as per the risk-mitigation activities.
The practical considerations to be taken into account while preparing a PER include:
- The reasoning behind the clinical evidence gathering methods used, including literature searches (related protocols and reports)
- A description of the technology behind the IVD
- The intended purpose and associated claims
- The actual scientific validity and the analytical and clinical performance data that have been evaluated
- The clinical evidence supporting the use of the device when assessed in the context of the current state-of-the-art
- Any new conclusions coming from post-market performance follow-up or other sources.
The impact of European In-Vitro Diagnostic Regulation 2017/746 (IVDR) on the device industry is more profound than the impact of European Medical Device Regulation 2017/745 (MDR). The majority of IVDs under the earlier IVD directives were self-certified and did not involve any notified bodies for conformity assessment. In contrast, around 90% of IVDs now require the involvement of notified bodies. Also, new requirements for establishing the performance of an IVD have been introduced in the EU IVDR, adding a significant volume of regulatory work for IVD manufacturers. Receipt of insufficient or irrelevant data will result in the issuance of major non-conformities by notified bodies. You should take into account all the practical considerations described above when building the performance reports for your IVDs.
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0746&rid=6
- https://ec.europa.eu/health/sites/default/files/md_sector/docs/md_mdcg_2020_1_guidance _clinic_eva_md_software_en.pdf
- https://ec.europa.eu/health/sites/default/files/md_sector/docs/md_mdcg_2020_guidance _classification_ivd-md_en.pdf
- https://ec.europa.eu/health/sites/default/files/md_sector/docs/mdcg_2021-4_en.pdf
- https://ec.europa.eu/health/sites/default/files/md_sector/docs/md_mdcg_2019_11_guidance _qualification_classification_software_en.pdf
About The Author:

Like what you are reading?
Sign up for our free newsletter, newsletter signup.

Get the latest articles from Med Device Online delivered to your inbox.
- CLINICAL EVALUATION REPORT
- CLINICAL EVALUATION PLAN
- CLINICAL DATA ANALYSIS
- LITERATURE SEARCH PROTOCOL
- DEMONSTRATION OF EQUIVALENCE
- MEDICAL DEVICE CLINICAL EVALUATION
- PERFORMANCE EVALUATION PLAN
- SCIENTIFIC VALIDITY REPORT
- CLINICAL TRIALS
- Scientific Validity Report
Scientific Validity Report IVDR

A scientific validity report is used to evaluate the performance of an IVD device, according to IVDR Regulation 2017/746. You must do this early in the life of your IVD if you are introducing a new product to the market. One approach to achieving this is to use the appropriate scientific validity information on instruments that measure the same analyte or marker.
You may and should, of course, carry out extensive searching of the literature; however, if there is insufficient data, you will have to do concept, pre-clinical, or clinical study proof. Your scientific validity report summarizes all the results, which will be included in your performance assessment report. For further information, see IVDR Annex XIII, Part A.
The Scientific Validity Report Services We Provide:
Identification and revision of the scientific validity claims in the intended purpose, systematic search and selection of peer-reviewed literature on each scientific validity claim, critical evaluation of the literature and writing of the scientific validity report, literature search plan, systematic literature search reporting, establishment of scientific validity, what is the procedure for producing a scientific validity report.
(a) Examine the IVD's stated function and the scientific claims that it makes.
(b) For each of the identified scientific validity claims, a systematic literature search of peer-reviewed literature and additional sources specified above was conducted.
(c) The findings of the literature search are screened, and the literature is evaluated.
(d) The report must be written in a systematic fashion once all relevant material has been obtained.
(e) The report's scientific validity is being compiled.
What are the Most Important Factors to Consider When Developing SVR?
(a)The analytical and clinical performance parts of the Performance Evaluation Report (PER) must be in accord with the scientific validity presented
(b) An IVD's scientific validity is not a one-time operation; it must be confirmed on a regular basis during its existence.
(c) The required literature search must be conducted using pre-defined literature search and selection techniques, which will be recorded in literature selection tables and assembled into a literature search and selection report.
(d) Cycle of revisions with the client to ensure that the scientific validity is properly established.
(e) Scientific validity will assure the benefit-risk ratio's continuous acceptance, as well as the capacity to predict developing dangers based on factual information.
(f) For gathering appropriate information, screening and evaluating data, and compiling an SVR, a mix of scientific, clinical, and regulatory expertise, as well as solid writing abilities, is essential.
What are the many sources of data used to compile SVR?
(a)Data on the scientific validity of devices that measure the same analyte or marker.
(b) Scientific literature (peer-reviewed)
(c) Expert opinions/positions from relevant professional groups that have reached a consensus
(d) Proof-of-concept studies' findings
(e) Clinical performance studies findings
Reduced burden for your employees
Contact us with your plans or ask us more information
Send Question
Performance Evaluation and Performance Studies of in Vitro Diagnostic Medical Devices Under the IVDR
- Living reference work entry
- First Online: 29 October 2022
- Cite this living reference work entry

- Wolfgang Ecker 4
Part of the book series: Reference Series in Biomedical Engineering ((TIENRE))
73 Accesses
IVDs are a backbone of modern state-of-the-art medicine, especially forthcoming precision and personalized medicine; its reliability is of paramount importance for most medical disciplines, including for medical genetic testing, prediction, staging or diagnosis of cancer, or proper microbiological diagnosis. This holds true for individual as well as public health relevant diagnosis and covers both professional and lay use. It is therefore important that the performance of IVDs (both analytical and clinical) is always based on sufficient clinical evidence which can be demonstrated throughout the life cycle of these products (e.g., to address emerging new variants of infectious agents).
Performance evaluation of IVDs must therefore be put high on the agenda of modern regulatory attempts in Europe and globally. The European legislator had to create a well-anchored and sophisticated valid life cycle process for performance evaluation, benchmarking, and even improving the global guidelines of the International Medical Device Regulators Forum (IMDRF). Apart from proper scientific literature search, performance studies of IVDs will be the major source of valid scientific data to underpin the clinical evidence needed for an IVD.
This is a preview of subscription content, log in via an institution to check access.
Access this chapter
Institutional subscriptions
Abbreviations
Unique Device Identifier – Device Identifier for identifying manufacturer and IVD model (unit of use)
Union-wide unique single identification number for performance studies of IVDs
Coordinating Member State
European Commission
Common Specification
Declaration of Interest by clinical evaluator(s)
Device Specific Guidance
Evidence-based Medicine
European Norm
European Databank for Medical Devices (and IVDs)
Food and Drug Administration, US
Global Harmonisation Task Force
General Safety and Performance Requirements, see IVDR, Annex I
Investigators Brochure
International Electrotechnical Commission, standardization body
International Medical Device Regulators Forum
International Standardization Organization
In vitro Diagnostic (medical device)
IVD-Regulation
Health Technology Assessment
Medical Device Coordination Group, issues MDCG Guidance
Medical Device Software
Medical Device Guideline by COM for the old Directives
Member State of EU/EEA
Notified Body, EU conformity assessment body
Performance Evaluation Plan
Performance Evaluation Report
Post Market Performance Follow-up
Post Marker Surveillance, by manufacturer
Performance Study Plan
Quality Management system
Single Registration Number (for EUDAMED)
Unique Device Identifier – Device Identifier
Unique Resource Locator (Internet)
European Parliament and European Council (2017a) Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU. Retrieved from: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02017R0746-20220128&from=DE ; cited as: IVDR 2017/746
European Parliament and European Council (2017b) Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 concerning medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 9s0/385/EEC and 93/42/EEC. Official Journal of the European Union. L 117, 1-175, cited as: MDR 2017/745. Retrieved from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745
European Parliament and European Council (2014) Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC, cited as CTR 536/2014. Retrieved from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32014R0536
Global Harmonization Task Force (2012a) Clinical evidence for IVD medical devices – Key Definitions and Concepts. Retrieved from: https://www.imdrf.org/working-groups/clinical-evidence-ivd-medical-devices
Global Harmonization Task Force (2012b) GHTF/SG5/N8:2012: Clinical evidence for IVD medical devices – clinical performance studies for in vitro diagnostic medical devices. Retrieved from: https://www.imdrf.org/sites/default/files/docs/ghtf/final/sg5/technical-docs/ghtf-sg5-n8-2012-clinical-performance-studies-ivd-medical-devices-121102.pdf
Global Harmonization Task Force (2012c) Global Harmonization Task Force (2012): GHTF SG5 scientific validity determination and performance evaluation. Retrieved from: https://www.imdrf.org/sites/default/files/docs/ghtf/final/sg5/technical-docs/ghtf-sg5-n7-2012-scientific-validity-determination-evaluation-121102.pdf
Medical Device Coordination Group (MDCG) (2022a) MDCG 2022–10: Q&A on the interface between Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR). Retrieved from: https://ec.europa.eu/health/document/download/59abcc81-fd32-4546-a340-24c8fad4e2ac_en?filename=mdcg_2022-10_en.pdf
Medical Device Coordination Group (MDCG) (2022b) MDCG 2022–9: summary of safety and performance template. Retrieved from: https://ec.europa.eu/health/document/download/b7cf356f-733f-4dce-9800-0933ff73622a_en?filename=mdcg_2022-9_en.pdf
Medical Device Coordination Group (MDCG) (2022c) MDCG 2022–2: guidance on general principles of clinical evidence for In Vitro Diagnostic medical devices (IVDs). Retrieved from: https://ec.europa.eu/health/document/download/f373538f-939c-472f-9536-436b6ddac085_en?filename=mdcg_2022-2_en.pdf
Medical Device Coordination Group (MDCG) (2021): MDCG 2021–21 Rev.1: guidance on performance evaluation of SARS-CoV-2 in vitro diagnostic medical devices. Retrieved from: https://ec.europa.eu/health/document/download/729f09dc-9f95-40b9-a62a-a0e9fff1d252_en?filename=mdcg_2021-21_en.pdf
Medical Device Coordination Group (MDCG) (2020) MDCG 2020–1: guidance on clinical evaluation (MDR)/Performance evaluation (IVDR) of medical device software. Retrieved from: https://ec.europa.eu/health/document/download/19d9e24f-2808-4e00-bfeb-75892047407d_en?filename=md_mdcg_2020_1_guidance_clinic_eva_md_software_en.pdf
Medtech Europe (2021) Clinical evidence requirements for CE certification under the In Vitro Diagnostic Regulation in the European Union, 2nd ed. Nov 2021. Retrieved from: https://www.medtecheurope.org/wp-content/uploads/2020/05/medtech-europe-performance-evaluation-second-edition-1.pdf
International Standardization Organization (ISO) (2019) ISO 20916:2019: in vitro diagnostic medical devices – clinical performance studies using specimens from human subjects – good study practice
Google Scholar
Download references
Author information
Authors and affiliations.
University of Applied Sciences Upper Austria for Medical Technology, Linz, Austria
Wolfgang Ecker
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Wolfgang Ecker .
Editor information
Editors and affiliations.
Institute of Health Care Engineering, Graz Univ of Technology, Graz, Austria
Christian Baumgartner
Human.technology Styria GmbH, Graz, Austria
Johann Harer
Institute of Health Care Engineering with European Testing Centre of Medical Devices, Graz University of Technology, Graz, Austria
Jörg Schröttner
Rights and permissions
Reprints and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this entry
Cite this entry.
Ecker, W. (2022). Performance Evaluation and Performance Studies of in Vitro Diagnostic Medical Devices Under the IVDR. In: Baumgartner, C., Harer , J., Schröttner, J. (eds) Medical Devices and In Vitro Diagnostics. Reference Series in Biomedical Engineering(). Springer, Cham. https://doi.org/10.1007/978-3-030-98743-5_9-1
Download citation
DOI : https://doi.org/10.1007/978-3-030-98743-5_9-1
Received : 03 July 2022
Accepted : 09 July 2022
Published : 29 October 2022
Publisher Name : Springer, Cham
Print ISBN : 978-3-030-98743-5
Online ISBN : 978-3-030-98743-5
eBook Packages : Springer Reference Engineering Reference Module Computer Science and Engineering
- Publish with us
Policies and ethics
- Find a journal
- Track your research
+44 (0)20 8996 7029
IVDR: Practical Considerations for the Performance Evaluation Plan and Report
- October 23, 2020
- Medical Devices
- Jacques du Preez

The IVDR (EU 2017/746) brings new requirements for manufacturers with regard to Performance Evaluation and Clinical Performance Studies and one of those is the need for a Performance Evaluation Plan (PEP) and Performance Evaluation Report (PER).
What is a PEP?
The PEP should be constructed to enable the evaluator to correctly perform and appropriately report on the Performance Evaluation. The PEP should cover at least the following (per the IVDR) or should include a justification for its exclusion:
- details of the intended purpose, or intended use, of the IVD;
- the performance specifications for the IVD, as established by the manufacturer to ensure that the intended purpose is fulfilled;
- the identity of the analyte or marker to be determined by the IVD;
- any related certified reference standards or measurement procedures to allow for metrological traceability;
- the clearly identified specific target patient groups for the IVD along with indications, limitations and contra-indications;
- the relevant GSPRs that the Performance Evaluation will support;
- details of the methodology, including appropriate statistical tools, to be used in the assessment of the analytical and clinical performance of the IVD and the limitations of the device and information provided by it;
- a description of the state of the art relating to the IVD and including any relevant standards, common specifications, guidance documents, etc.;
- the parameters to be used (based on the state of the art in medicine) to determine the acceptability of the benefit-risk ratio for the intended purpose/s and for the analytical and clinical performance of the IVD;
- an overview of each of the development phases for the IVD with milestones, acceptance criteria and timing of verification and validation testing;
- if the IVD is software used as a medical device, then details of reference databases and other sources of data used as the basis for the device’s decision making;
- the process for PMPF planning.
Taking time to prepare the PEP with sufficient detail – as with any experimental protocol or research plan – will improve both efficiency and effectiveness of the Performance Evaluation process when it is conducted. It is likely that a number of stakeholders may need to be engaged to develop, and then implement, the PEP. For smaller companies, some of these may not be immediately available within the organisation and careful consideration of appropriate and timely resourcing should also be part of this Performance Evaluation planning process.
W hat is a PER?
The PER has its roots in the Clinical Evaluation Report (CER) required under the Medical Device Directive (MDD) and now the Medical Device Regulation (MDR). The PER should be an output from the process of Performance Evaluation as noted in previous blog posts and be populated from the results of applying the Performance Evaluation Plan.
Annex VIII, Part A (1.3.2) of the IVDR outlines the specific components of the PER and specify that it must include:
- the scientific validity report;
- the analytical performance report;
- the clinical performance report; and
- an assessment of those reports supporting the demonstration of the clinical evidence as sufficient to make a decision on the benefit-risk ratio.
Other aspects that the PER should cover are the reasoning behind the clinical evidence gathering methods used including any literature searches (and their related protocols and reports); a description of the technology behind the IVD; the intended purpose and associated claims; the actual scientific validity and the analytical and clinical performance data that has been evaluated; the clinical evidence supporting the use of the device when assessed in the context of the current state of the art in medicine; and any new conclusions coming from PMPF or other sources.
This means that the PER is likely to contain all of the device related analytics such as sensitivity, specificity, reproducibility, stability, and so on. Those companies that have prior experience with FDA 510(k) submissions for their devices may already have some elements o fthe PER in place. However, as the PER must support the clinical benefit of the IVD test, there will inevitably be data that will needed to be generated and gathered as appropriate.
Performance Evaluation Reports for Class C and D devices must be updated at least annually, whereas PERs for Class A and B devices should be updated as needed, although at least a three years review cycle would be recommended.
Practical Considerations
From the above it can be seen that there are a number of key aspects that should be considered in the Performance Evaluation process, but in particular the three areas of scientific validity, analytical performance and clinical performance.
In each of these areas, whether the devices in question are already on the market or those in development, the manufacturer should begin by developing a gap analysis that will answer some basic questions, such as:
What technology is the device based on and how does it compare to the current state of the art of scientific knowledge?
- Is this research by the manufacturer, by an academic institution, by an independent evaluator, etc.?
- Have proof of concept studies been conducted to the right level of good laboratory practice and are the results sufficiently robust?
- What standards or references have been used for these tests?
- Do the results provide a high level of assurance?
Based on the answers to these and other questions specific to the IVD and intended performance in question, the manufacturer will be able to develop a detailed picture that will inform the Performance Evaluation Plan. Alongside this technical gap analysis and action plan the manufacturer should also develop a detailed financial feasibility analysis to ensure that the correct levels of resources are available at the appropriate time for the Performance Evaluation and associated activities such as clinical data gathering by means of a Clinical Performance Study. The latter will be discussed in more detail in a future blog post.
For manufacturers with IVDs already on the market or those with device in development, time is short. The Date of Application (May 2022) is a hard deadline for these devices to have achieved their certification under the IVDR. Therefore, sufficient time needs to be factored in for on-site QMS audit to the IVDR, and reviews of required technical documentation. Bearing in mind that there are a significant volume of these devices on the market, and the capacity of NBs will not be infinite, it would seem prudent to allow at least 12 months from application to certification. Therefore, it is vital that a manufacturer starts this process as soon as possible to ensure all appropriate additional testing and data may be gathered in sufficient time. So don’t wait – if you haven’t already started this process, start now!
Author: Jacques du Preez, COO and Robin Stephens, CEO, Psephos Biomedica
The Compliance Navigator blog is issued for information only. It does not constitute an official or agreed position of BSI Standards Ltd or of the BSI Notified Body. The views expressed are entirely those of the authors.

How to Establish the State-of-the-Art Within the Scientific Validity Report
In this practical presentation, Criterion Edge’s Principal Medical Writer, Dr. Sarah Chavez, presents focused strategies on building an IVDR-aligned SOA section within the SVR. She also talks about the critical role of the SVR as it relates to the overall performance evaluation process. In addition, you will learn how to assess your SVR project for unanticipated roadblocks and delays, and how indecision or incomplete supporting documents can negatively impact SOA (and therefore, SVR) quality and timelines.
Who should watch this session?
Those Regulatory, Quality and Clinical leaders, regulatory writers, and teams who are tasked with the development, writing, review or approval of Performance Evaluation Reports for EU IVDR submission, or anyone interested in learning more about IVDR requirements for PERs.
Sign Up to Watch
To watch the free webinar recording, How to Establish the State-of-the-Art Within the Scientific Validity Report , fill out the form below.
Are you a medical writer? Yes No
Are you interested in working for Criterion Edge? Yes No
Medical Writing Experience CER CSR/Annual Report Protocol Risk Management Plan eCTD Modules/Submission documents IB/IFU Clinical Narratives Non-Clinical Reports Safety/Pharmacovigilance Other
Therapeutic areas of experience Cardiovascular Oncology Endocrine/Metabolic Orthopedic Vaccines/Biologics Gastroenterology/Hepatology Opthamology/ENT Renal Neurology/Neuromodulation
What industry do you currently work in? Medical Device Pharmaceuticals In Vitro Diagnostics (IVD) Other Medical Other Non-Medical
Key Takeaways:
- The foundational role of the SOA in the SVR
- The required components of the SOA such as the safety and performance objectives
- The importance of a robust systematic literature review (SLR) to support your SOA
- +32 (0)14 490 422
Scientific validity reports
Based on in depth literature review
- Scientific Validity Reports
As per IVD-Regulation 2017/746, the performance evaluation of an IVD Medical Device consists of three pillars:
- Analytical Performance Studies
- Clinical Performance Studies
Qarad can provide you with the complete Performance Evaluation documentation package or one or multiple parts.
Click here for more information about the Analytical and Clinical performance Studies.
Analytical and Clinical Performance Studies
What is Scientific Validity?
Scientific validity is concerned with the association of the analyte of a device with one or more clinical conditions or physiological states (Regulation 2017/746, Art. 2, definition 38). This association is a fundamental factor in the justification process for development and production of IVD devices.
At Qarad, we can provide you with scientific validity reports that follow the procedures as stipulated by the IVD Regulation (Annex XIII, 1.2.1). The service is entirely based on a systematic literature study and involves:
- Identification and revision of the scientific validity claims for the intended purpose of the IVD device, in collaboration with the customer
- Systematic search and selection of peer-reviewed literature on each scientific validity claim
- Critical evaluation of the literature and writing of the scientific validity report
- Literature search and selection protocol
- Literature selection tables
- Literature search and selection report
- Revision cycle with the customer to further guarantee that the scientific validity is adequately established

Experience with Scientific Validity Report writing
We have created Scientific Validity Reports for many reputable companies before.
See our fact overview below.
Regulatory Experts in IVD
Qarad has many years of experience in European IVD Regulations, which allows us to understand the bigger picture.
Proven expertise

We can provide you with a complete documentation package with your performance evaluation:
- Scientific Validity Report
- Clinical performance report
- Analytical performance report

Personal approach
We follow your specific demands and are in constant interaction to fulfill your needs.

Independence
We make sure that the quality and content of the Scientific Validity Reports is consistent and does not depend on the individual visions of each expert within the company.

Reduced burden for your employees
Avoid overwhelming your employees with the excessive work which comes with the pressing demands of the new IVD regulation.
- Qarad has already written more than 100 Scientific Validity Reports, for very diverse markets.
- We have 9 customers who have outsourced their Scientific Validity Report writing to us.
- Four of those customers belong to the Top 10 IVD companies.
Contact us for more information
If you decline, your information won’t be tracked when you visit this website. A single cookie will be used in your browser to remember your preference not to be tracked.

Navigating IVDR Compliance: Applicability of Scientific Validity for IVDs
While established over ten years ago as a concept by the precursor to the International Medical Device Regulators Forum (IMDRF), the Global Harmonization Task Force (GHTF), scientific validity is a novel concept in the EU regulatory framework in the transition from the IVDD to the IVDR.
Understanding scientific validity for IVDs
The first step in establishing the scientific validity for an IVD or determining if scientific validity is applicable to an IVD is understanding exactly what ‘scientific validity’ means for the specific IVD of interest.
While the IVDR defines ‘scientific validity of an analyte’ as ‘the association of an analyte with a clinical condition or a physiological condition’, the following definitions are also established in the respective sources indicated:
- The extent to which the analyte, or marker to be determined by the IVD is associated with the targeted physiological state or clinical condition ( MDCG 2022-2 )
- For IVD medical device software (MDSW): The extent to which the claimed intended purpose is associated with the targeted physiological state or clinical condition (derived from MDCG 2022-2 )
- For IVD MDSW: The association of an MDSW output with a clinical condition or physiological state ( MDCG 2020-1 )
While these definitions are (relatively) easily applied to reagent-based IVDs (including reagent-containing kits) and standalone MDSW, they are less easily applied to other types of IVDs.
Applicability of scientific validity for Class A devices under Rule 5(b) of IVDR
Certain Class A devices, such as those that fall under Rule 5(a) (e.g. products for general laboratory use intended by the manufacturer to make them suitable for in vitro diagnostic procedures relating to a specific examination) or Rule 5(c) (i.e. specimen receptacles) do not perform analyte or marker determination and are not IVD MDSW. Therefore, it is reasonable for manufacturers of such IVDs to justify the non-applicability of scientific validity.
However, in the case of Class A devices that fall under Rule 5(b) (i.e. instruments intended by the manufacturer specifically to be used in vitro diagnostic procedures), critical considerations regarding the applicability of scientific validity include:
- Does the instrument determine an analyte or marker without the use of any additional reagents? If the answer is yes, in all likelihood the instrument is in fact not a Class A IVD based upon guidance provided in MDCG 2020-16 , and scientific validity would be applicable unless due justification is provided.
- Does the instrument incorporate IVD MDSW?
Identification of applicable software models and scientific validity for IVD MDSW
For the second consideration, it’s important to clearly identify which of the software models described in MDCG 2020-1 is applicable, i.e.
- MDSW with an intended purpose and claimed clinical benefit related to driving or influencing a medical device for a medical purpose, in which case scientific validity would be applicable and would need to encompass the MDSW and the driven or influenced instrument.
NOTE: If the MDSW is intended to drive/influence more than one IVD, an independent scientific validity report (and performance evaluation) is required for each foreseen and clinically viable software-device combination.
- Software driving or influencing the use of a medical device with no independent intended purpose or independent claimed clinical benefit, in which case scientific validity would be applicable to the instrument including the software as either a component or accessory.
Applicability of scientific validity for calibrators and control materials in IVDs
Other types of IVDs for which the applicability of scientific validity is not clearly established are calibrators and control materials, as they do not determine an analyte or marker but are actually themselves measured by the IVD that they are intended to calibrate or serve as a control. MDCG 2022-2 notes that “…for certain devices intended to be used together, for example a reagent intended to be used with calibrators and controls, it may be more appropriate to establish the scientific validity in the context of this combination.” In our experience, Notified Bodies expectations during performance evaluation review of standalone calibrators and control material is that these include a scientific validity report demonstrating the association of the analyte or marker for the assay(s) they are supporting (all analytes/markers in the case of multiparametric controls/calibrators).
Types and sources of evidence for establishing scientific validity in IVDs
Once a manufacturer has determined the type and applicability of scientific validity to their device, the next step is to determine the type/level of evidence necessary to support scientific validity which will largely be driven by the IVDs degree of novelty. Annex XIII of the IVDR establishes that manufacturers shall demonstrate scientific validity based on one or a combination of the following sources:
- Relevant information on the scientific validity of devices measuring the same analyte or marker.
- Scientific (peer-reviewed) literature.
- Consensus expert opinions/positions from relevant professional associations.
- Results from proof-of-concept studies.
In the case of well-established scientific validity , for which the generally acknowledged state of the art has been characterized, manufacturers will typically establish scientific validity based upon data from options 1-3 due to the existence of high-quality evidence in the public domain (refer to our article on best practices for performance evaluation literature searches here ). For novel devices, or where manufacturers are unable to identify sufficient data from options 1-3, a scientific rationale will need to be provided and the manufacturer will need to generate new or additional data, in most cases from options 4-5.
Establishing a literature search protocol and report for IVD clinical performance
As scientific peer-reviewed literature is also among the data sources to demonstrate IVD clinical performance, and Section 1.2, Annex XIII of the IVDR states that as a general methodological principle, manufacturers shall identify through a systematic scientific literature review the available data relevant to the device and its intended purpose and identify any remaining unaddressed issues or gaps in the data, both a literature search protocol and report including scientific validity among their objectives should be established. Only once this literature search has been completed and any gaps identified and addressed should manufacturers then proceed with compilation of the scientific validity report (it is recommended that the literature search protocol and report be included as annexes to the performance evaluation report as it will typically support scientific validity, clinical performance and state of the art data analyzed within the scope of the performance evaluation).
Learn more with MedEnvoy
If you have any additional questions regarding performance evaluation or require relevant training / consulting services, get in touch .
Explore similar topics
- Best Practices for Conducting Literature Searches in IVDR Peformance Evaluation
- Understanding the Specific Evaluation Documentation Required for IVD Performance Under Article 56 of the IVDR
- Planning Performance Evaluation Under the IVDR
- Analytical Performance Characteristics for IVDs: What Manufacturers Need to Know
You also might be interested in

Shipping Devices and IVDs Directly to Consumers? | EU MDR Economic Operators
The EU MDR/IVDR establishes specific regulatory obligations for economic operators,[...]

Medical Device Registration in India
The Central Drug Standard Organization (CDSCO), which falls under the[...]
Swiss IvDO: Fast-Approaching Deadlines for IVD Manufacturers
As we’ve previously reported for non-IVD devices, under the new[...]
© 2024 · MedEnvoy, Global BV – The Hague, The Netherlands. Privacy Policy | Contact Us
Type and press Enter to search
Clinical evidence requirements according to the IVDR 2017/746: practical tools and references for underpinning clinical evidence of IVD-MDs
Affiliations.
- 1 Université de Franche-Comté, LINC, CIC 1431 INSERM, Centre Hospitalier Universitaire de Besançon, Besançon, France.
- 2 Tech4Health Network, FCRIN, Besançon, France.
- PMID: 36919280
- DOI: 10.1515/cclm-2022-1252
In May 2022, the European Regulation 2017/746 (IVDR) came into force. It changes the approach of in vitro medical devices (IVD-MDs) for industry and institutions. It reinforces the clinical evidence requirements to improve performance, safety and transparency. Despite extended transition periods and existing guides, IVDR remains difficult to interpret and bringing devices into compliance requires efforts. The generation of clinical evidence is essential to demonstrate compliance with IVDR, and encompasses scientific validity, analytical performance and clinical performance. It is required to demonstrate, per intended use in the target population and clinical care pathway, IVD-MDs clinical performance (compared to a predefined clinical performance). Thus, there is a need for IVD-manufacturers and end-users in health care institutions, to obtain guidance on how to generate this clinical evidence. This article aims industrials and clinicians to identify key steps imposed by the IVDR for bringing IVD-MDs to the EU-market. We propose a general view of performance evaluation requirements for IVD-MDs and provide key references, including how to establish study design that will enable to document clinical performance of existing, refined or emerging medical tests. Finally, we propose a roadmap to address the relevant questions and studies in relation to the documents requested in the IVDR.
Keywords: in vitro diagnostic; 2017/746 IVDR; analytical performance; clinical evidence; clinical performance; performance evaluation.
© 2023 the author(s), published by De Gruyter, Berlin/Boston.
- Equipment and Supplies* / standards
- European Union
- Government Regulation*
Book a Meeting
Ctd conversion.
In June 2010, The Medicines Control Council (MCC) announced the intention to implement the South African Common Technical Document (ZA CTD) format which will replace the current MRF1 and any applications still in MBR1 format.
From June 2011, submissions in ZA CTD format are mandatory (excluding veterinary medicines).
Freyr is currently working with many Global Pharmaceutical and Consumer Health Care companies in supporting them in planning and executing the CTD conversion requirement for the existing and new product registrations in South Africa enabling them to meet the MCC mandate. For some of these global companies CTD conversion is a time consuming and a huge responsibility that needs careful planning and execution given their growing product portfolio in the African market.
Drop us a URL
Ebook pdf download, get started now, freyr’s global presence.
North America
- Canada - English
- USA - English
- Mexico - English
South America
- Argentina - English
- Bolivia - English
- Brazil - English
- Chile - English
- Colombia - English
- Costa Rica - English
- Dominican Republic - English
- Ecuador - English
- French Guiana - English
- Guatemala - English
- Guyana - English
- Panama - English
- Paraguay - English
- Peru - English
- Puerto Rico - English
- Suriname - English
- Uruguay - English
- Venezuela - English
East Europe
- Kazakhstan - English
- Kosovo - English
- Kyrgyzstan - English
- Serbia - English
- Tajikistan - English
- Turkmenistan - English
- Ukraine - English
- Uzbekistan - English
- Albania - English
- Andorra - English
- Armenia - English
- Austria - English
- Belarus - English
- Belgium - English
- Bosnia & Herzegovina - English
- Bulgaria - English
- Croatia - English
- Cyprus - English
- Czech Republic - English
- Denmark - English
- Estonia - English
- Finland - English
- France - English
- Georgia - English
- Germany - German
- Greece - English
- Hungary - English
- Iceland - English
- Ireland - English
- Italy - English
- Latvia - English
- Liechtenstein - English
- Lithuania - English
- Luxembourg - English
- Macedonia - English
- Malta - English
- Moldova - English
- Monaco - English
- Montenegro - English
- Netherlands - English
- Norway - English
- Poland - English
- Portugal - English
- Romania - English
- San Marino - English
- Slovakia - English
- Slovenia - English
- Spain - English
- Sweden - English
- Switzerland - English
- United Kingdom - English
- Vatican City - English
North Africa & Middle East
- Algeria - English
- Bahrain - English
- Benin - English
- Burkina Faso - English
- Burundi - English
- Cameroon - English
- Central African Republic - English
- Chad - English
- Djibouti - English
- Egypt - English
- Eritrea - English
- Ethiopia - English
- Gabon - English
- Gambia - English
- Ghana - English
- Guinea - English
- Guinea Bissau - English
- Iraq - English
- Israel - English
- Ivory Coast - English
- Jordan - English
- Kenya - English
- Kuwait - English
- Lebanon - English
- Liberia - English
- Libya - English
- Mali - English
- Mauritania - English
- Morocco - English
- Niger - English
- Nigeria - English
- Oman - English
- Palestine - English
- Qatar - English
- Rwanda - English
- Saudi - English
- Senegal - English
- Sierra Leone - English
- Syria - English
- Togo - English
- Tunisia - English
- Turkey - English
- United Arab Emirates - English
- Uganda - English
- Yemen - English
Southern Africa
- Angola - English
- Botswana - English
- Democratic Republic of the Congo - English
- Lesotho - English
- Madagascar - English
- Mauritius - English
- Malawi - English
- Mozambique - English
- Namibia - English
- Seychelles - English
- South Africa - English
- Swaziland - English
- Tanzania - English
- Zambia - English
- Zimbabwe - English
- Bangladesh - English
- Bhutan - English
- Brunei - English
- Cambodia - English
- China - English
- Fiji - English
- Hong Kong - English
- India - English
- Indonesia - English
- Laos - English
- Malaysia - English
- Maldives - English
- Mongolia - English
- Myanmar - English
- Nepal - English
- Pakistan - English
- Philippines - English
- Russia - English
- Singapore - English
- Sri Lanka - English
- Taiwan - English
- Thailand - English
- Timor Leste - English
- Vietnam - English
Australia & New Zealand
- Australia - English
- New Zealand - English
Japan & South Korea
- Japan - English
- South Korea - English
Good Manufacturing Practice
Good Manufacturing Practice is that part of Quality Assurance which ensures that products are consistently produced and controlled to the quality standards.
- Regular periodic or rolling quality reviews of all registered medicinal products, including export only products are conducted.
- The Manufacturer and Holder of Certificate of Registration, where different, should evaluate the results of the review and an assessment should be made of whether corrective and preventative action or any revalidation should be undertaken.
Medicines Control Council’s (MCC) general policy is that the standard to be used to assess compliance with current Good Manufacturing Practice (cGMP), is the South African Guide to Good Manufacturing Practice (SA guide to GMP).
- Under Section 22C of the Act, all South African manufacturers should be licensed
- The Act requires that overseas manufacturers of medicine supplied to South Africa should comply with the same or equivalent manufacturing standards as expected of South African manufacturers
- When acceptable evidence of GMP compliance is not available, overseas manufacturers are inspected by the GMP Inspectorate before registration of the medicine is approved
Let Us Source
Medicines registration.
Medicines to be used in South Africa for both public and private sectors shall be duly registered with the national regulatory authority, the Medicines Control Council’s (MCC) in accordance with the provisions and requirements of the Medicines and Related Substances Control Act No. 101 of 1965 and the Regulations and Guidelines published in terms thereof.
A written notification from the Minister to the effect that the medicine is considered essential to national health; an expert report (which is not more than 2 (two) years old; a package insert (where the product has been approved) and a summary basis for the registration (SBRA) should be submitted with application.
The Registrar shall notify the applicant within 30 days of the date of receipt of the application and the Council shall, within 9 months make a decision with regard to the application.
The abbreviated medicine review process is based mainly on the expert reports of the pharmaco toxicological and clinical data.
Applications for Abbreviated Medicine Review Process (AMRP) can only be accepted if the product has been approved by the said authorities within the last three years of the license in the licensing country.
Partner With Us
Please fill the form to download, please fill the form to download white paper.
Regulatory Operations
Submissions publishing.
In June 2010, The Medicines Control Council (MCC) announced the intention to implement the South African common technical document (ZA CTD) format which will replace the current MRF1 and any applications still in MBR1 format. From June 2011, submissions in ZA CTD format are mandatory (excluding veterinary medicines).
With the need for converting old format submissions to CTD format comes the opportunity for Market Authorization Holders (MAH) to plan and be ready for electronic submissions. Freyr can compile submissions in eCTD format and print in paper format as required by the current MCC requirement. This allows the MAHs to be prepared for future eCTD requirements from MCC and enables efficient electronic submission dossier management.
Registrations and Submissions Information Management
Module 1 (Administrative 1.10 Foreign Regulatory Status) requirement from Medicines Control Council’s (MCC) requires the applicant to provide a list of countries in which an application for the same product is being applied for in South Africa has been submitted, dates of submission (if available). This should detail approvals (with indications). Applicants must declare whether a marketing application for the medicine has been rejected in the countries listed under 1.10.1 prior to submission of the application in South Africa. If the medicine has been rejected, repeatedly deferred or withdrawn, then the MCC must be informed and the reasons supplied.
If no application has been submitted for registration in the country of origin, include a statement to provide the reason for this decision. It should be noted that aforementioned information is required to be provided in dossier however, it does not mean that this will help to speed up the review process.
Request a Demo
SVR stands for Scientific Validity Report, a mandatory document for an In-Vitro Diagnostic (IVD) Device. SVR features the association of an analyte with a clinical condition or a physiological state. The association justifies the whole process of an IVD development and production, to demonstrate general safety and performance of an IVD.
What Legal Basis Does an SVR Require?
The requirements for the Scientific Validity Report (SVR) are defined in In-Vitro Diagnostic Regulations (EU) 2017/746; definition 38 of Article 2 defines the Scientific Validity and definition 48 of the same article defines the Performance Evaluation. The performance evaluation of an IVD is defined as, “assessment and analysis of data to establish or verify the scientific validity, the analytical and, where applicable, the clinical performance of a device”.
What are the Key Considerations for Developing SVR?
There are various aspects to be considered while developing an SVR, such as:
- The scientific validity demonstrated shall be in line with the analytical and clinical performance sections of the Performance Evaluation Report (PER)
- The scientific validity of an IVD is not a one-time activity and requires continuous confirmation throughout the lifecycle of an IVD
- Required literature search shall be carried out based on pre-defined literature search and selection protocols, captured in literature selection tables and compiled in literature search and selection report
- Revision cycle with the customer to further guarantee that the scientific validity is adequately established
- Scientific validity shall ensure continued acceptability of the benefit-risk ratio and of detecting emerging risks on the basis of factual evidence
- A combination of scientific, clinical and Regulatory knowledge along with good writing skills are required for collecting necessary information, screening and evaluation of data followed by compilation of an SVR
What are the Different Sources of Information for Compiling SVR?
The manufacturer shall demonstrate scientific validity of an IVD based on the information available from various sources, such as:
- Relevant information on the scientific validity of devices measuring the same analyte or marker
- Scientific (peer-reviewed) literature
- Consensus expert opinions/positions from relevant professional associations
- Results from proof-of-concept studies
- Results from clinical performance studies
The validity of the analyte or marker inferenced from the above sources should be demonstrated and documented in the scientific validity report.
What is the Step-By-Step Process for Compiling SVR?
The gathering of necessary information and compilation of a Scientific Validity Report involves a systemic step-by-step process as detailed below:
- Evaluate intended purpose of an IVD and determine the valid scientific claims
- Systemic literature search of peer-reviewed literature and other sources listed above for each of the determined scientific validity claim
- Screening of the literature search results, evaluation of the literature
- Once all the relevant information is gathered, the report shall be developed in a structured format
- Compilation of the scientific validity report
As a part of Performance Evaluation Report (PER) services, Freyr delivers EU IVDR compliant Scientific Validity Reports as a standalone service. To know how Freyr assisted a French-based client to submit high-quality SVRs to Notified Bodies, in a short span of one month, download the proven Case here . Would you like to know more about Freyr’s specialized expertise in SVR? Reach us at [email protected] for comprehensive insights.
- New Product Authorization - Finished Products
- New Market Authorizations - APIs
- Post Approval - CMC and Life Cycle Management
- Regulatory Assessment | Gap Analysis
- Regulatory Consulting and Strategy
- Health Authority Queries – Responses | Interactions
- Market Authorization Application (MAA) - Article 10 (1)
- MAA - Post-Approval Submissions
- MAA - License Renewal
- Investigational Medicinal Product Dossiers (IMPD)
- Biosimilars MAA – Article 10(4)
- Active Substance Master File (ASMF) Submissions
- Certification of Suitability (CEP) Submissions
- Sister CEP Submissions
- Brexit-related Regulatory Support
- Dossier Templates for USFDA and EU Regulatory Submissions
- Product, Market & Regulatory Pathway Strategy
- Strategic Regulatory Services – Africa
- Regulatory Strategy for Emerging Markets
- Drugs Regulatory Intelligence
- Medical Devices Regulatory Intelligence
- Nutraceuticals Regulatory Intelligence
- Regulatory Market Intelligence Reports
- Health Authority Directives
- Regulatory Intelligence
- Global eCTD Publishing and Submission Services
- NeeS Submissions
- Paper Submissions
- Legacy Conversions
- CSR - Report Level Publishing
- Structure Product Labeling/Monograph (SPL-SPM) Submissions
- Regulated Product Submissions (RPS)
- Drug Master File (DMF) Submissions
- Regulatory/Medical Review of Ad Promo Material
- Clinical Labeling
- Global Labeling
- TGA’s New Product Information (PI) Format
- Labeling Compliance
- Medical Devices Labeling
- Food/Dietary Supplements Labeling
- Packaging Artwork Process Consulting
- Artwork Lifecycle Coordination
- Artwork Management Staffing Services
- Artwork Management System PLM Tool
- Artwork Proofreading / Quality Check
- Artwork RA Labeling
- Artwork Studio
- Content to Carton
- Food and Food Supplements
- Medical Devices
- Regulatory Writing Services
- Clinical Trial and Consulting Services
- Clinical Trial Audit and Monitoring Services
- Nonclinical Regulatory Writing
- Permitted Daily Exposure (PDE)
- Toxicological Risk Assessment (TRA)
- Scientific Writing
- Manuscript Writing and End-to-End Services
- ICSR Services
- Literature Monitoring Services
- Aggregate Safety Reports
- Signal Management
- Risk Management
- PV Audit and Training
- Pharmacovigilance Safety Database
- Medical Device PMS/Vigilance
- Regulatory Intelligence and Consulting in Pharmacovigilance
- Quality Control
- GxP Audit Services
- SOP Writing and SOP Review Services
- Computer System Validation and Computer System Assurance
- End-to-End Compliance Services
- Remote and Virtual Audit
- Spin-offs, Mergers & Acquisitions, and Divestitures (SoMAD) Consulting services
- Pharmaceuticals
- Innovator Drugs
- Biosimilars
- Over-The-Counter Drugs
- Czech Republic
- Netherlands
- New Zealand
- Saudi Arabia
- European Authorized Representative (EAR)
- US Agent Service
- Authorized Representative
- Labeling & Promotional Material Review
- Post Market Surveillance Support
- QMS Compliance Services
- Regulatory Compliance, Gap Analysis & Remediation
- Regulatory Strategy
- Regulatory Submission and Registration
- Translation
- 510(k) Submission Process Consulting
- 513(g) Submission Process Consulting
- Switzerland
- Safety Assessment and Toxicology
- Formulation Review and Product Classification
- Cosmetics Claims and Substantiation
- Labeling Review
- Cosmetics Packaging Artwork Solutions
- Product Information / Technical File Compilation
- Nutraceuticals Claims Review
- Product Labeling Review
- Technical Documents/ Dossier Compilation
- Freyr iREADY - Ready to Use Ingredients Database
- Kingdom of Saudi Arabia
- Philippines
- South Korea
- United Kingdom
- Freyr SUBMIT PRO
- Freyr iREADY
- Freyr IMPACT
- Freyr SPL-SPM
- Quality Assurance
- Support and Maintenance
- Global <span>Presence</span>
- End-to-end <span>Regulatory Services</span>
Scientific Validity Reports
Ivd regulation 2017/746 introduces many new challenges. one of the most remarkable is the introduction of scientific validity..
- What is clinical evidence?
- What is Scientific Validity?
- How to prove Scientific Validity?
Clinical evidence
While the old IVD directive mainly focused on the provision of analytical performance data and limited clinical performance data, IVD Regulation 2017/746 entails more elaborate requirements in terms of safety and performance with the introduction of the concept ‘clinical evidence’. Clinical evidence consists of clinical data and performance evaluation results, pertaining to a device of sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s) when used as intended by the manufacturer. Confirmation of the conformity of a device with the applicable general safety and performance requirements, in particular those concerning the performance characteristics, shall be based on:
- Scientific Validity
- Analytical Performance
- Clinical Performance

Scientific validity
As defined in the regulation, Scientific Validity means "the association of an analyte with a clinical condition or physiological state".
To demonstrate the scientific validity, a manufacturer will need to use one or a combination of the following sources:
- relevant information on the scientific validity of devices measuring the same analyte or marker
- scientific (peer-reviewed) literature identified through a systematic literature review
- expert opinions/positions from relevant professional associations
- results from proof of concept studies
- results from clinical performance studies
All the information collected during this process, should be clearly structured and well-documented in a Scientific Validity Report.

Advantages of outsourcing scientific validity report writing
Writing scientific validity reports based on systematic literature review requires a lot of time and effort and should follow a sound procedure. Moreover, the authors of such reports should possess substantial scientific knowledge, good writing skills and he/she should understand the regulatory background to make sure the report fulfils all requirements.

Ready to roll?
In the best interest of your company, you should outsource your scientific validity reports to external experts. A good regulatory expert who also has extensive experience in the field of IVDs can assist you in meeting this new regulatory requirement with minimal impact on your already stretched resources.
IMAGES
VIDEO
COMMENTS
The IVDR outlines that evidence for an IVD's conformity is established by demonstrating and substantiating the scientific validity, analytical performance and clinical performance. Furthermore, the IVDR underlines that the necessary clinical evidence should be based on a sufficient amount and quality of data in order to allow
essential to establish scientific validity and clinical performance, support compliance, and ensure patient safety. We will also discuss the newly introduced Scientific Validity Report (SVR) as an integral part of the total documentation to be submitted under the regulation. 2. Key changes from IVDD to IVDR
Clinical Evidence for IVD medical devices - Scientific Validity and Performance Evaluation Study Group 5 Final Document GHTF/SG5/N7:2012 November 2nd, 2012 Page 5 of 20 how to appraise and analyze the data; and the content of the clinical evidence report. The guidance contained within this document applies to devices that meet the definition of an
Scientific Validity is a crucial component of the clinical evidence required under the IVDR. It provides the clinical background and context for the environment in which the IVD operates and is necessary to determine performance criteria against which to compare the IVD under evaluation. A methodical, detailed and documented approach to the ...
"Assessment and analysis of data to establish or verify the scientific validity, the analytical and, where applicable, the clinical performance of a device" IVDR Article 2(44) This definition uses other defined terms: Scientific validity; Analytical performance; Clinical performance; Definition: Scientific validity of an analyte
The performance evaluation report is an output of the process of performance evaluation activities populated from the results of applying the performance evaluation plan. Annex XIII, Part A (1.3.2) of the IVDR outlines the specific components of the PER and specifies that it must include: The scientific validity report.
Scientific validity is demonstrated and documented in the scientific validity report. Analytical Performance "Analytical performance" means the ability of a device to correctly detect or measure a particular analyte (IVDR 2017/746, Art. 2 (40)). Only if the IVD measures as exactly as possible, what it is supposed to measure
A scientific validity report is used to evaluate the performance of an IVD device, according to IVDR Regulation 2017/746. You must do this early in the life of your IVD if you are introducing a new product to the market. One approach to achieving this is to use the appropriate scientific validity information on instruments that measure the same ...
Scientific validity is demonstrated and documented in the scientific validity report. 2.3.2.2 Analytical Performance " Analytical performance " means the ability of a device to correctly detect or measure a particular analyte (IVDR 2017/746, Art. 2 (40)).
Back to Blog Listing. Under the EU In Vitro Diagnostic Medical Devices Regulation 2017/746 (IVDR), every IVD must have a Performance Evaluation Report (PER). PERs consist of three pillars: scientific validity, analytical performance and clinical performance. Collating data to satisfactorily address each pillar must be a priority for IVD ...
Other aspects that the PER should cover are the reasoning behind the clinical evidence gathering methods used including any literature searches (and their related protocols and reports); a description of the technology behind the IVD; the intended purpose and associated claims; the actual scientific validity and the analytical and clinical ...
Key Takeaways: The foundational role of the SOA in the SVR. The required components of the SOA such as the safety and performance objectives. The importance of a robust systematic literature review (SLR) to support your SOA. This webinar outlines the steps to build an IVDR-aligned State-of-the-Art section within the Scientific Validity Report.
What is Scientific Validity? Scientific validity is concerned with the association of the analyte of a device with one or more clinical conditions or physiological states (Regulation 2017/746, Art. 2, definition 38). This association is a fundamental factor in the justification process for development and production of IVD devices.
Your scientific validity report will summarise your findings and become part of your performance evaluation report. For further information, please see IVDR Annex XIII, Part A. Evaluating Analytical Performance of your IVD. IVDR Annex I, Section 9.1 comprehensively describes the performance characteristics that must be measured. Said ...
With a robust plan to follow and analyses of scientific validity, analytic, and clinical performance completed, it's time to assemble the final performance evaluation report. Here's where all that time you spent planning pays off. Annex XIII, Part A, 1.3.2 of the IVDR spells out the basic requirements for your PER including:
Scientific validity - IVDR defines scientific validity as "the association of an analyte with a clinical condition or physiological state." For example, hemoglobin is correlated with anemia and other blood disorders. In this component of the PE, you will be expected to justify the use of any analytes, markers, or targets you use and ...
The Performance Evaluation Report contains the methods and results regarding scientific validity, analytical performance and clinical performance. There's a separate standard available for that: EN 13612:2002. It's very short and doesn't contain a whole lot of information. Additionally, there are three IMDRF guidance documents: GHTF/SG5 ...
While the IVDR defines 'scientific validity of an analyte' as 'the association of an analyte with a clinical condition or a physiological condition', the following definitions are also established in the respective sources indicated: ... an independent scientific validity report (and performance evaluation) is required for each foreseen ...
The generation of clinical evidence is essential to demonstrate compliance with IVDR, and encompasses scientific validity, analytical performance and clinical performance. It is required to demonstrate, per intended use in the target population and clinical care pathway, IVD-MDs clinical performance (compared to a predefined clinical ...
The requirements for the Scientific Validity Report (SVR) are defined in In-Vitro Diagnostic Regulations (EU) 2017/746; definition 38 of Article 2 defines the Scientific Validity and definition 48 of the same article defines the Performance Evaluation. ... Freyr delivers EU IVDR compliant Scientific Validity Reports as a standalone service. To ...
In this practical presentation, Criterion Edge presents focused strategies on building an IVDR-aligned State of the Art (SOA) section within the Scientific Validity Report (SVR). We also talk about the critical role of the SVR as it relates to the overall performance evaluation process. Key Takeaways: The foundational role of the SOA in the SVR
Scientific validity. As defined in the regulation, Scientific Validity means "the association of an analyte with a clinical condition or physiological state". To demonstrate the scientific validity, a manufacturer will need to use one or a combination of the following sources: All the information collected during this process, should be clearly ...
In this practical presentation, Criterion Edge presents focused strategies on building an IVDR-aligned State of the Art (SOA) section within the Scientific Validity Report (SVR). We also talk about the critical role of the SVR as it relates to the overall performance evaluation process. Key Takeaways: